
AutoGAMESS

If you use AutoGAMESS for any published research, please cite it in the following or similar manner:
AutoGAMESS[1] was used for workflow automation.
[1] Ferrari, Brian. "AutoGAMESS: A Python package for automation of GAMESS (US) Raman calculations." Journal of Open Source Software 4.41 (2019): 1612.
This is a python module for automating the generation of input files and parsing of log files with end goal of generating Raman data using the GAMESS(us) Quantum Chemistry software.
This package was developed using GAMESS VERSION = 20 APR 2017 (R1), it has also been partially tested for use with GAMESS VERSION = 1 MAY 2013 (R1) and GAMESS VERSION = 14 FEB 2018 (R1).
AutoGAMESS is also able to generate line plots of vibrational frequency vs. IR/Raman intensities. Generated plots will be titled with the molecule name in the file and the theory/basis set used for the calculation. Each symmetry group will be plotted in a different color, from either a default or user specified color list. The spectral line (sum of line broadening) will also be plotted in red with 50% transparency. An example is shown bellow using Lorentzian line broadening method with the default sigma option.
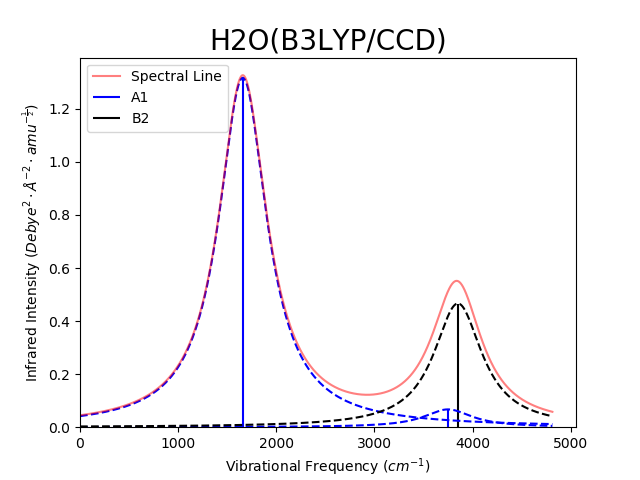 | 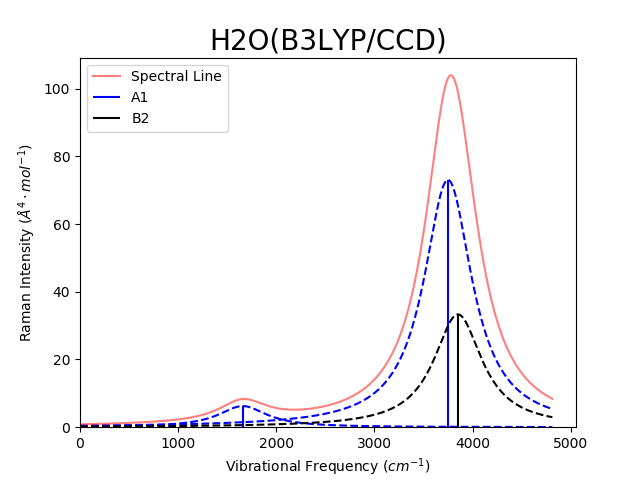 |
Installing AutoGAMESS
AutoGAMESS can be installed using
python -m pip install autogamess --user
AutoGAMESS requires all the following Python packages:
- Python3.x
- NumPy
- SciPy
- Pandas
- basis_set_exchange
- PeriodicElements
- openpyxl
- Matplotlib
Tests
AutoGAMESS uses pytest for testing, from within the tests directory execute the following to run tests.
python -m pytest
Function Documentations
new_project
new_project(maindir,csvfile,initial_coords_dict=None,title='Project_Name/', make_inputs=False)
This function creates a new directory tree for a GAMESS project, also makes
a couple of text files for use with other functions.
Parameters
----------
maindir: string
A directory string (including the final `/`) that points to the
directory that the project tree will be spawned in.
csvfile: string
A directory string (including the final `.csv`) that points to the
text file containing project information. Read module documentation
for csv file format.
initial_coords_dict: dictionary [Optional]
This should be a dictionary with the key being the specie and the
value being a list that of its initial coordinates.
title: string [Optional]
A directory string (including the final `/`) that will be used as
the head of project directory tree.
make_inputs: boolean True/False [Optional]
if True then new_project will call input_builder at the end.
Notes 1
----------
If the molecules you wish to build are not already defined in the
general autogamess coordinate dictionary, then initial_coords_dict
must be passed.
To see the autogamess coordinate dictionary simply print out
>>> ag.dictionaries.molecule_dictionary
Returns
----------
This function returns nothing
Notes 2
----------
The format of the spawned directory tree is as follows:
maindir
|
title
-------------------------------------
| | | | |
Codes Inps Logs Batch_Files Spreadsheets
| | | |
--------- Block ----------- 1 file per specie
| | | | |
Text_Files Scripts Fail Pass Sorted
| | |
------- Block 1 directory per specie
| |
Unsolved Solved
Sections in directory tree labeled 'Block' are directory trees with the
following format:
1 directory per run type
|
1 directory per specie
Examples
----------
>>> import autogamess as ag
>>>
>>> csvfile = './input.csv'
>>> maindir = './'
>>> title = 'Project Title/'
>>>
>>> ag.new_project(maindir, csvfile, title=title)
>>>
input_builder
input_builder(inputfile, save_dir, initial_coords_dict=None,proj_title=' Your Title Goes Here\n')
This function builds optimization input files.
Parameters
----------
inputfile: string
This should be a full directory string that points to the input
csv file.
save_dir: string
this should be a full directory string that points to the directory
you wish to save the inputs in.
initial_coords_dict: dictionary [Optional]
This should be a dictionary with the key being the specie and the
value being a list of the symmetry group and symmetry unique atom
coordinates. Examples can be seen in the AutoGAMESS GitHub repository
as well as by prinint out the default dictionary, see Notes section.
proj_title: string [Optional]
This should be a string ending with `\n`
Notes 1
----------
If the molecules you wish to build are not already defined in the
general autogamess coordinate dictionary, then initial_coords_dict
must be passed.
To see the autogamess coordinate dictionary simply print out
>>> ag.dictionaries.molecule_dictionary
Returns
----------
This function returns nothing
Notes 2
----------
This function uses the EMSL Basis Set Exchange module to import
external basis sets[1]. This function also uses the Periodic_Elements
package by VaasuDevanS [2].
[1] https://github.com/MolSSI-BSE/basis_set_exchange
[2] https://github.com/VaasuDevanS/Periodic_Elements
Examples
----------
>>> import autogamess as ag
>>>
>>> csvfile = './input.csv'
>>> savedir = './'
>>> title = 'Project\n'
>>>
>>> ag.input_builder(csvfile, savedir, proj_title=title)
>>>
opt2hes
opt2hes(optfile, logfile)
This function writes a hessian calculation input file using a previously
run optimization input file and the log file generated by the calculation.
Parameters
----------
optfile: string
This should be a string that points to the input file of an
already run optimization file. (FULL DIRECTORY STRING REQUIRED)
logfile: string
This should be a string that points to the log file of an
already run optimization file. (FULL DIRECTORY STRING REQUIRED)
Returns
-------
This function returns nothing if it terminates successfully, otherwise
it returns ValueError.
Example
-------
>>> import autogamess as ag
>>>
>>> logfile = './Optimization_Log_Folder/IBv6_NH3_CCSD-T_CC6_opt.log'
>>> optfile = './IBv6_NH3_CCSD-T_CC6_opt.inp'
>>>
>>> ag.opt2hes(optfile, logfile)
>>>
hes2raman
hes2raman(hesfile, datfile)
This function writes a raman calculation input file using a previously
run hessian input file and the dat file generated by the calculation.
Parameters
----------
hesfile: string
This should be a string that points to the input file of an
already run hessian file. (FULL DIRECTORY STRING REQUIRED)
datfile: string
This should be a string that points to the DAT file of an
already run hessian file. (FULL DIRECTORY STRING REQUIRED)
Returns
-------
This function returns nothing if it terminates successfully, otherwise
it returns ValueError.
Example
-------
>>> import autogamess as ag
>>>
>>> datfile = '../restart/IBv6_NH3_CCSD-T_CC6_hes.dat'
>>> hesfile = './IBv6_NH3_CCSD-T_CC6_hes.inp'
>>>
>>> ag.hes2raman(hesfile, datfile)
>>>
sort_logs
sort_logs(projdir, logsdir)
This function sorts all the loose log files in the 'Logs' directory.
Parameters
----------
projdir: string
A directory string (including the final `/`) that points to the
project head directory.
logsdir: string
A directory string (including the final `/`) that points to the
directory containing the log files.
Returns
----------
This function returns nothing
Notes
----------
For this function to work properly the project directory tree must be
in the exact format that the 'new_project' function spawned it in.
Examples
----------
>>>import autogamess as ag
>>>
>>>projdir = './Example/'
>>>logsdir = './logs/'
>>>
>>>ag.sort_logs(projdir, logsdir)
>>>
fill_spreadsheets
fill_spreadsheets(projdir=False, sorteddir=False, sheetsdir=False)
This function fills in the spreadsheets initially generated by new_project
with data collected from the outfiles of calculations.
Parameters
----------
projdir: string [Optional]
This should be a full directory string pointing to the project
directory initially created by new_project.
sorteddir: string [Optional]
This should be a full directory string pointing to the sorted log
files directory.
sheetsdir: string [Optional]
This should be a full directory string pointing to the spreadsheets
directory.
Notes
----------
If projdir is not passed to fill_spreadsheets function then both other
parameters MUST be passed to it. Similarly if projdir is passed the
other two parameters MUST be left blank.
Once `fill_spreadsheets` has parsed the data file, it will move the log file
into the `Pass` directory implying that the calculation successfully terminated, or it will move it to `Fail` directory if termination unsuccessful.
Returns
-------
This function returns nothing.
Example
-------
>>> import autogamess as ag
>>>
>>> projdir = './Your Project Title/'
>>>
>>> ag.fill_spreadsheets(projdir)
>>>
>>> import autogamess as ag
>>>
>>> sorteddir = './project/Logs/Sorted/'
>>> sheetsdir = './project/Spreadsheets/'
>>>
>>> ag.fill_spreadsheets(sorteddir=sorteddir, sheetsdir=sheetsdir)
>>>
get_data
get_data(filename)
This function collects data from GAMESS(us) log files.
Parameters
----------
filename: string
This should be a string that points to the log file of any
GAMESS(us) calculation. (FULL DIRECTORY STRING REQUIRED)
Returns
-------
data: object
This is an object with all the data collected from the log file.
Below is a list of the attributes associated with `data` based on
each log file type.
all files: `cpu`, `time`
opt files: `bond_lengths`, `bond_angles`
hes files: `vib_freq`, `ir_inten`
raman files: `raman`
vscf files: `vscf_freq`, vscf_ir
Notes
-------
This function is primarily intended for interal use by AutoGAMESS.
Example
-------
>>> import autogamess as ag
>>>
>>> filename = './AGv0-0-6_NH3_CCSD-T_CC6_opt.log'
>>>
>>> ag.get_data(filename)
>>>
make_plot
make_plot(file, savedir=None, cmap=['b', 'k', 'r', 'g', 'y', 'c'], method=None, sig=300, flag=[], reverse_x=True)
This function make vibrational frequency vs. IR/Raman intensity line plots.
Parameters
----------
file: string
This should be a string that points to the log file of a hessian or
Raman GAMESS(us) calculation. (FULL DIRECTORY STRING REQUIRED)
savedir: string
This should be a string that points to the directory in which you
would like to save the png of the plot.(FULL DIRECTORY STRING REQUIRED)
cmap: list [Optional]
This should be a list of Matplotlib allowed color choices. Each symmetry
will be plotted with a different color in the list.
method: string [Optional]
This should be string giving the method for line broadening, options are
`Gaussian`, `Lorentzian`, None(defualt).
sig: integer or float [Optional]
This should be a numerical value to be used as the FWHM for the line
broadening method chosen. Default: 300 wavenumbers
flag: list [Optional]
This should be a list of integers, in particular 1,2 and 3. This list
tells the function what to plot and what to omit from the plot.
Please see the Notes section for more details.
reverse_x: boolean True/False [Optional]
if True then x-axis will be in reverse (ie: 300---150----0).
Notes
-------
The `flag` parameter is used as follows:
[Default] `flag=[]` ---> All lines are plotted
`flag=[1]` ---> Vertical lines are not plotted
`flag=[2]` ---> Spectral line not plotted
`flag=[3]` ---> Gaussian/Lorentzian lines not plotted
`flag=[1,2]` --->V Vertical lines and Spectral line not plotted
List combination follow the same format, all possible list combinations
are allowed.
Returns
-------
This function returns nothing.
Example
-------
>>> import autogamess as ag
>>>
>>> file = './AGv0-0-6_NH3_CCSD-T_CC6_hes.log'
>>> savedir = './'
>>>
>>> ag.make_plot(file, savedir)
>>>
>>> cmap = ['b', 'r', 'k', 'c']
>>> ag.make_plot(file, savedir, cmap=cmap)
>>>
>>> method = 'Lorentzian'
>>> sig = 450
>>>
>>> ag.make_plot(file, savedir, cmap=cmap, method=method, sig=sig)
>>>
generate_scaling_factors
generate_scaling_factors(projdir, expt_dict, species, method='scott')
This function generates scaling factors and scaled frequencies.
Parameters
----------
projdir: string
This should be a full directory string pointing to the project
directory initlly created by new_project.
expt_dict: dictionary
This should be a python dictionary with the experimental frequency
values for all species that the user wants to generate scaling factors
for in it. Format is explained in Notes section.
species: list
This should be a list of all species the user would like scaling factors
generated for. Any molecule in the list must have experimental data in
the `expt_dict` associated with it.
method: string [Optional]
This should be string giving the method for scaling factor calculation,
options are `scott`(defualt).
Notes
-------
`expt_dict` format should be as follows:
{`specie`: [`nu_1`, `nu_2`, ... , `nu_N`]}
where `specie` must be written the same way as the Excel spreadsheet file
for that molecule is written. Each frequency, `nu`, should be given in
the same order as they appear (left to right) in the spreadsheet.
`species` list format can be in any order but must adhere to the rule
that any element in `species` is a key for `expt_dict`
Once execution of this function is completed the `Hessian` worksheet
will be updated to have a coulmn giving `Scaling Factor/RMS`, as well
as the scaled frequencies will appear in parathesis next to the predicted
frequencies.
Returns
-------
This function returns nothing.
Example
-------
>>> import autogamess as ag
>>>
>>> projdir = './Your Project Title/'
>>> expt_dict = {'H2O': [1595, 3657, 3756]}
>>> species = ['H2O']
>>>
>>> ag.generate_scaling_factors(projdir, expt_dict, species)
>>>
Input Descriptions
All user functions contain doc strings with examples and explanations of parameters and returns. However, a few functions require specific inputs not fully explained in the doc strings. Such as the functions:
- new_project
- input_builder
The CSV file required by both functions must have the following format. The first line must be the header, written exactly as follows.
All lines after the header should give input as 1 item per column per line. As shown in the example bellow.
| H2O | B3LYP | G32CCSD | CCD | may-cc-pVQZ | Optimization |
| NH3 | MP2 | G4MP2 | CCT | aug-cc-pV7Z | Hessian |
| HCN | CCSD-T | G4MP2-6X | CCQ | may-cc-pVTZ | Raman |
| H2CO | PBE | CCCA-S4 | CC5 | Sadlej-pVTZ | VSCF |
| CH4 | wB97X-D | CCCA-CCL | CC6 | jun-cc-pVQZ | |
| C2H6 | SCS-MP2 | ACCD | jul-cc-pVTZ | ||
| C2H4 | CCSD2-T | ACCT | |||
| C2H2 | ACCQ |
AutoGAMESS assumes the user will be performing every possible combination of Theory and Basis Sets(internal and external) for every calculation type, across all species. Therefore repetition within columns will cause an error. If a user wishes to perform Optimization, Hessian and Raman calculations on water(H2O) using only B3LYP CCD the following should be in the CSV.
| H2O | B3LYP | CCD | Optimization | ||
| Hessian | |||||
| Raman |
For any given CSV file the total generated input files by input_builder will be n*m*i where n is the number of Species given, m is the number of Theory given, and i is the number of Basis Sets given.
Internal basis sets should be written in the same format as they are required by GAMESS(us) inputs.
External basis sets should be written in the same format as they as required by ESML basis_set_exchange. Some external basis sets have shorthand names built into AutoGAMESS to prevent special characters such as (, ), +, etc. from being put into file names. Notice this is applicable to may-cc-pV(D+d)Z written simply as may-cc-pVDZ similarly for the other calendar basis sets.
initial_coords_dict is another input parameter that requires specific formatting. The dictionary is meant to give the initial guess coordinates for a particular symmetry of a molecule. This should be a python dictionary that has the Species (molecule) name as the key and a list with the following format.
key = 'H2O'
value = ['CnV 2,\n','\n',
' O 8.0 -0.0000000000 0.0000000000 -0.0123155409\n',
' H 1.0 -0.0000000000 -0.7568005555 0.5926935705\n']
initial_coords_dict = {key : value}
Note that the atoms given in the list are the symmetry unique atoms for the point group symmetry given in the first element of the list. Point group symmtery should be given in GAMESS(us) format, with the second element of the list being \n for cases where GAMESS(us) requires a blank card after the symmetry group. For symmetry groups that cannot have a blank card after in GAMESS(us) the second element should be the first symmetry unique atom. Finally, make sure all elements of the list end with \n to ensure they are written in separates lines.
Some molecules are already compiled within AutoGAMESS default dictionary however, if one of the molecules in the input CSV file is not within the default dictionary a complete dictionary with all molecules within the CSV file is required by AutoGAMESS.
Examples of Common AutoGAMESS Utilization Methods
Compile Existing Data
A basic script for generating a new project directory, sorting already existing logs into it, then filling the spreadsheets with the data in the existing output files. For this script to work properly, file names must adhere to the AutoGAMESS file naming convention
[arbitrary thing]_[Specie]_[Theory Level]_[Basis Set]_[Abbreviated Run Type].[inp/log/dat]
An example is “AG-test_H2O_B3LYP_CCD_opt.log”, where the Arbitrary Thing is AG-test, Specie is H2O, Theory Level is B3LYP, Basis Set is CCD and the Abbreviated Run Type is opt.
The ‘arbitrary thing’ section can be anything, since this is typically where AutoGAMESS will write the version number. Since AutoGAMESS reads information from file names and requires the underscore separates the information something must be present there to prevent confusion. If the file name format is incorrect the fill_spreadsheets function will be unable to map the data to the correct cell in the spreadsheet. However, the get_data function only requires the abbreviated run type be written with underscore before and file extensive following it (ie: ..._opt.log). The Abbreviated Run Types are,
Optimization = opt
Hessian = hes
Raman = raman
VSCF = vscf
import autogamess as ag
maindir = './'
csvfile = './input.csv'
title = 'Project Title/'
ag.new_project(maindir, csvfile, title=title)
projdir = maindir + title
logsdir = './Logs/'
ag.sort_logs(projdir, logsdir)
ag.fill_spreadsheets(projdir)
Converting Input Files
A basic script for converting all files within a directory into their next calculation type. Also separates the files that GAMESS(us) calculation did not terminate successfully.
import os
import autogamess as ag
idir = './inps/'
ldir = './logs-dats/'
done = './done/'
fail = './failed/'
iext = '.inp'
lext = '.log'
dext = '.dat'
for file in os.listdir(ldir):
if lext not in file:
continue
if '_opt' in file:
inp = idir + file.replace(lext, iext)
dat = ldir + file.replace(lext, dext)
log = ldir + file
try:
ag.opt2hes(inp, log)
except:
os.rename(inp, inp.replace(idir, fail))
os.rename(log, log.replace(ldir, fail))
os.rename(dat, dat.replace(ldir, fail))
continue
os.rename(inp, inp.replace(idir, done))
os.rename(log, log.replace(ldir, done))
os.rename(dat, dat.replace(ldir, done))
if '_hes' in file:
inp = idir + file.replace(lext, iext)
dat = ldir + file.replace(lext, dext)
log = ldir + file
try:
ag.hes2raman(inp, dat)
except:
os.rename(inp, inp.replace(idir, fail))
os.rename(log, log.replace(ldir, fail))
os.rename(dat, dat.replace(ldir, fail))
continue
os.rename(inp, inp.replace(idir, done))
os.rename(log, log.replace(ldir, done))
os.rename(dat, dat.replace(ldir, done))
Data Parsing Log Files
A less common method of utilizing AutoGAMESS is to parse any single output file for data. The get_data function which is typically meant to be an internally used function can be called by the user. This will retrieve the data from the file, it will read the file name to get the run type.
import autogamess as ag
file = 'AG-test_H2O_B3LYP_CCD_opt.log'
data = ag.get_data(file)
lengths = data.bond_lengths
angles = data.bond_angles
Generating Scaling Factors
To generate scaling factors for all Hessian calculations that have been compiled by fill_spreadsheets you just need to call generate_scaling_factors. Here is an example considering the molecule in question is H2O.
import autogamess as ag
projdir = './Your Project Title/'
expt_dict = {'H2O': [1595, 3657, 3756]}
species = ['H2O']
ag.generate_scaling_factors(projdir, expt_dict, species)
Making Plots
Make plots with AutoGAMESS is quite simple, here are some examples (these examples are also found in the make_plot doc string).
This first example is to for having a plot displayed on screen without saving it.
import autogamess as ag
file = 'AG-test_H2O_B3LYP_CCD_opt.log'
ag.make_plot(file)
The next example shows how to make and save a plot to the current working directory.
import autogamess as ag
file = './AGv0-0-6_NH3_CCSD-T_CC6_hes.log'
savedir = './'
ag.make_plot(file, savedir)
The next example shows you how to pick your own colors for each symmetry group that is plotted.
import autogamess as ag
file = './AGv0-0-6_NH3_CCSD-T_CC6_hes.log'
savedir = './'
cmap = ['b', 'r', 'k', 'c']
ag.make_plot(file, savedir, cmap=cmap)
The next example shows how to use line broadening in your plot, in particular this uses the Lorentzian method and a Full Width Half Maximum (FWHM) of 450 wavenumbers.
import autogamess as ag
file = './AGv0-0-6_NH3_CCSD-T_CC6_hes.log'
savedir = './'
cmap = ['b', 'r', 'k', 'c']
method = 'Lorentzian'
sig = 450
ag.make_plot(file, savedir, cmap=cmap, method=method, sig=sig)
The next example shows how to use flags to omit certain things from being plotted. Here we are omitting the vertical lines and the dashed liner broadening lines, leaving only the spectral line to be plotted.
import autogamess as ag
file = './AGv0-0-6_NH3_CCSD-T_CC6_hes.log'
savedir = './'
cmap = ['b', 'r', 'k', 'c']
method = 'Lorentzian'
sig = 450
flag = [1,3]
ag.make_plot(file, savedir, cmap=cmap, method=method, sig=sig)